
Болезнь рихтера что это такое
Содержание:
Лимфопролиферативные заболевания, особенно В-клеточные лейкозы – вялотекущие. Чаще при них используется тактика «наблюдай и жди». У некоторых пациентов эта патология может осложниться синдромом Рихтера. По каким-либо причинам параллельно имеющемуся лимфолейкозу у пациентов развивается вторая диффузная крупноклеточная опухоль. Проявляется патология ухудшением общего состояний, появлением симптомов развития крупноклеточной опухоли. В большинстве случаев синдром Рихтера ухудшает прогноз для жизни пациента, даже при использовании адекватной химиотерапии.
Причины развития синдрома Рихтера
Впервые случай возникновения крупноклеточной диффузной опухоли у пациента с хроническим лимфолейкозом был описан Рихтером ещё в 1928 году. До сих пор достоверно не известно, является ли возникшая опухоль клоном первичной, или же это два независимо возникших злокачественных образования. По данным иммунологических и генетических исследований, в 1/3 случаев диффузная крупноклеточная опухоль развивается из клональных клеток зрелоклеточной лимфомы, а в 2/3 случаев их взаимосвязь не доказана. Соответственно и точной причины, почему у некоторых пациентов развивается вторая опухоль, достоверно не известно. Считается, что предрасполагающими факторами являются:
- Вирус. При исследовании крупноклеточных лимфом у пациентов с синдромом Рихтера было выявлено ДНК вируса Эпштейна-Барра, являющегося предрасполагающим фактором развития и других типов лимфом.
- Иммуносупрессия из-за интенсивной химеотерапии. Частота развития синдрома Рихтера у больных хроническим лимфолейкозом увеличивается при приёме флударабина. При приёме пуриновых аналогов такая закономерность не обнаружена.
- Прогрессирование лимфопролиферативного заболевания. Некоторые исследователи считают, что синдром Рихтера является следствием развития хронического лимфолейкоза. Возникает у пациентов с индивидуальной предрасположенностью к такому осложнению.
Чтобы подтвердить или опровергнуть эти причины развития синдрома Рихтера, проведённых исследований недостаточно.
К тому же встречается синдром Рихтера крайне редко. Он развивается у 3-10 % пациентов со зрелоклеточными лимфопролиферативными патологиями. Появление крупноклеточной лимфомы ухудшает состояние пациента и сопровождается определёнными признаками.
Симптомы патологии
Чаще синдром Рихтера развивается на поздних стадиях и значительно осложняет течение хронического лимфолейкоза. Он проявляется:
- массивной лимфаденопатией;
- быстрым увеличением лимфоузла.
У некоторых пациентов по мере развития крупноклеточной диффузной лимфомы исчезает основной признак лимфолейкоза – увеличенное количество лимфоцитов в крови. У других же наоборот, лимфоцитоз значительно возрастает.
Помимо этого у больных проявляются симптомы опухолевой интоксикации и признаки ослабления гуморального иммунитета. Пациенты жалуются:
- на обильное потоотделение ночью;
- интенсивное беспричинное похудание;
- повышенную температуру тела;
- частые простуды;
- постоянно возникающие инфекционные заболевания мочевых путей;
- длительно заживающие кожные раны.
Боль возникает там, где расположены поражённые лимфоузлы.
Осложняется течение заболевания, если в патологический процесс вовлекается костный мозг и другие органы кроветворения. Нередко при синдроме Рихтера врач выявляет увеличенную селезёнку. И тогда пациенты жалуются на боль, давящее ощущение в брюшной полости.
Чтобы поставить точный диагноз, необходимы анализ крови, биопсия лимфоузла. Для гистологического исследования материал берут из наиболее подозрительных участков. Забор биоптата проводят с различных мест.
Прогноз при синдроме Рихтера
Дальнейшее прогрессирование болезни зависит от тактики лечения и индивидуальных особенностей пациентов. Часть пациентов после появления крупноклеточной лимфосаркомы умирают в течение полугода, даже если использовать адекватные для агрессивных лимфом комбинированные методы терапии.
Не всегда синдром Рихтера приводит к скорому летальному исходу. Особенно если самочувствие не изменяется. В этом случае продолжительность жизни при синдроме Рихтера значительно варьирует. Пациент может прожить от 3,5 месяцев до 9 лет.
При синдроме Рихтера пациенты нуждаются:
- в паллиативном лечении;
- спленэктомии (при значительно увеличенной селезёнке);
- комбинированном лечении.
При подборе схемы химеотерапии учитывают тип лимфом. Нередко цитостатики, цитотоксины дополняют противовоспалительными препаратами.
Пациентам с лимфомами, особенно при развитии синдрома Рихтера, необходимо избегать инфекционных заболеваний. Часто причиной смерти становится не сама опухоль, а неспособность иммунитета противостоять болезнетворным микроорганизмам.
К какому врачу обратиться
Синдром Рихтера развивается у пациентов с лимфопролиферативными заболеваниями. Такие больные находятся под наблюдением у врачей-гематологов, онкогематологов. Следует периодически проходить необходимые исследования, а при ухудшении самочувствия обязательно обратиться к лечащему врачу, чтобы вовремя выявить синдром Рихтера и изменить тактику лечения.

Болезнь Рейтера – аутоиммунное заболевание аллергического типа, сопровождается уретритом, конъюнктивитом и полиартритом. Вторичное патологическое состояние развивается после перенесенных инфекционных заболеваний кишечника и мочеполовой системы. Лечение синдрома Рейтера антибактериальными, противовоспалительными и антигистаминными препаратами ведет к полному выздоровлению, изредка процесс переходит в хроническое течение.

Этиология
Первые случаи синдрома Рейтера были описаны в начале прошлого столетия немецким военным доктором, по фамилии которого и названо заболевание. После перенесенной кишечной инфекции среди военнослужащих были зафиксированы случаи с одновременным поражением глаз, мочевой системы и суставов. Именно это состояние и назвали — болезнь Рейтера.
Развивается патология чаще у мужчин в возрасте до 35 лет после заражения венерическими заболеваниями с генетической предрасположенностью. Зафиксированы единичные случаи у детей и людей старшего возраста. Почти каждый из заболевших был носителем антигена HLA B27. Специфический белок локализуется на поверхности иммунных клеток, является одним из звеньев, обеспечивающих иммунные реакции.
У носителей антигена повышается риск заболеть серонегативными спондилоартритами, к которым относят болезнь Бехтерева, синдром Рейтера, острый увеит, ювенильный артрит. С другой стороны антиген защищает от таких патологий, как герпес, гепатит С, ВИЧ — инфекция.
Среди здорового населения выявляется до 10% антигеноносителей, встречаются случаи приобретенного состояния при приеме лекарственных препаратов – Сульфалазин, Каптоприл, Пеницилламин.
Классификация синдрома Рейтера
В медицинской практике различаются две формы болезни Рейтера:
- Спорадическая – развивается после венерических заболеваний, вызываемых хламидиями. Состояние появляется у отдельных людей, не имеет массового характера.
- Эпидемическая – появляется после сальмонеллеза, дизентерии, кишечного иерсиниоза. Патология возникает чаще в теплое время года, когда возможны эпидемии кишечных инфекций.
По длительности лечения течение болезни можно разделить на острое, которое продолжается до полугода и хроническое, не ограниченное по длительности.

Характерные симптомы
Признаки болезни Рейтера можно разделить на три группы, проявляющиеся друг за другом или одновременно:
- Уретрит – воспаление внутренних стенок мочевого канала. Симптомами являются рези и болезненное мочеиспускание, выделения гнойного характера. Если во время не начато лечение, к воспалению уретры присоединяется простатит.
- Конъюнктивит – аллергическое поражение слизистой оболочки глаз, представленное воспалением конъюнктивы. У больного чешутся и краснеют глаза, появляется слезотечение, небольшой отек век. Реакция распространяется на оба глаза, проходит в течение недели без специфического лечения.
- Артрит – поражение суставов может появиться не сразу. Появление симптома отмечается спустя неделю или месяц. Воспалительный процесс сопровождается интенсивными болями. Артрит поражает несколько сочленений, происходит это постепенно. Вначале болеть начинают суставы верхнего пояса, затем патология спускается на нижерасположенные соединения костей.
Суставное воспаление сопровождается синовитом. Пораженный участок отекает незначительно, в суставе скапливается умеренное количество патогенного содержимого. Отмечаются случаи общей интоксикации с появлением лихорадочного состояния.
При этом страдает двигательная функция, при длительном течении патология поражает сначала верхние конечности, затем нижние, после чего человеку становится тяжело передвигаться самостоятельно.
К основной триаде можно добавить поражение слизистой оболочки слизистой полости рта. Во рту появляются эрозивные гиперемированные язвы. Больной не может нормально принимать пищу, острая болезненность доставляет неприятные ощущения. Одним из частых осложнений во время течения болезни Рейтера является баланит (воспаление головки полового члена).
Дополнительными патологиями со стороны глаз может стать кератит и ирит. При кератите воспаляется роговица, синдром проявляется помутнением, появлением мелких язв на глазу и сильной болью. Ирит характеризуется воспалительными процессами на сосудистой оболочке глазного яблока. У человека резко снижается зрения, частое помутнение в глазах, наблюдается светобоязнь и слезотечение.
На теле появляются красные пятна, преимущественно на ладонях и подошвах стоп. Постепенно участки ороговеют, приводя к кератодермии. Со стороны сердечно-сосудистого тракта страдает сердечная мышца. Наблюдается развитие миокардита и миокардиодистрофии.
Стадии заболевания
Существуют две стадии болезни Рейтера:
- Инфекционная – начальная стадия появляется сразу после возникновения очага инфекции в мочеполовой системе. Период характеризуется восприимчивостью к лечению непосредственно против возбудителя инфекции. Если в ранний период добиться ликвидации очага воспаления, десенсибилизирующий контрагент прекратит свое влияние и выздоровление наступит быстрее.
- Иммунная – затяжной процесс приводит к развитию хронического иммунного ответа организма на длительное присутствие аллергена. На этом этапе развитие болезни не связано с первичной инфекцией, избавление от возбудителя не приносит положительного результата для лечения синдрома.
Причины появления
Почему появляется синдром Рейтера, выяснялось не один год учеными медиками. Основными провоцирующими факторами являются:
- перенесенные патологии, передающиеся половым путем, возбудителем которых является хламидия;
- присутствие хламидии в организме, доказано, что скрытые формы не проявляются, и человек может не подозревать, что внутри паразитирует вредоносный микроорганизм;
- кишечная группа инфекций – дифтерия, сальмонеллез, эширехиозы;
- наличие в организме антигена HLA B27, у предыдущих поколений может отмечаться случаи таких заболеваний, как псориаз, ревматоидные и ювенильные артриты, чаще всего крестцово-повздошных сочленений.

Диагностика синдрома Рейтера
Для подтверждения диагноза больной направляется к ряду медицинских специалистов: урологу, окулисту, ортопеду, ревматологу, иммунологу. Из лабораторных исследований назначают:
- лабораторное исследование крови: повышенная скорость оседания лейкоцитов и их увеличенное количество укажет на наличие воспаления;
- мазок из половых органов – наличие хламидий;
- при пункции из сустава проводится анализ жидкости на выявление возбудителя;
- биохимическое исследование крови на наличие ревматоидного фактора;
- анализ мочи по Нечипоренко подтверждает воспаление в уретре;
- определение антихламидийных антител в сыворотке крови;
- иммунологическое исследование крови на определение антигеноносительства;
- электрокардиограмма при нарушении работы сердца;
- рентгенография для обследования суставов;
- ультразвуковое исследование для определения количества и характера образовавшегося выпота в суставной сумке.
Терапевтические меры
При развитии острой фазы лечение болезни Рейтера направлено на устранение очага инфекции и воздействие на возбудителя:
- Антибиотики, активные по отношению к хламидии, купируют инфекционный процесс, уничтожая возбудителя. Спирамицин, Азитромицин, Левофлоксацин – одни из часто назначаемых препаратов, выписывать препараты может только врач, консультация обязательна! Данная группа препаратов вызывает нарушение микрофлоры кишечника. Для профилактики этого состояния назначается параллельный прием пробиотиков – Линекс, Нормабакт.
- Нестероидные противовоспалительные средства для снятия воспаления и обезболивающего эффекта – Индометацин, Кеторолак, Диклофенак.
- Антигистаминные препараты (Тавегил, Супрастин, Астемизол) блокируют аллерген, избавляют от проявлений десенсибилизации организма.
- Дезинтоксикационные средства для очищения крови и выведения токсинов из организма, применяются в форме диффузных растворов для внутривенного введения – Гемодез, Реополиглюкин, Полиамин.
- Витаминные комплексы для укрепления организма.
Лечение первой стадии не требует госпитализации, прогноз всегда благоприятный, больной быстро идет на поправку. При хроническом течении болезни Рейтера назначается стационарное лечение.
При этом проводится:
- Внутривенная антибиотикотерапия (Джозамицин, Ампициллин, Азитромицин) для антибактериального эффекта.
- Иммунносупрессоры – лекарственные средства, угнетающие иммунитет. Такролимус, Отесла, Ксолар – защищают организм от губительного воздействия иммунных клеток на собственный организм.
- Противоаллергическое лечение с помощью антигистаминных препаратов – Цетиризин, Меклизин, Астемизол.
- При длительном течении полиартрита назначается лечебная пункция с выкачиванием экссудата и терапевтическим промыванием растворами глюкокортикостероидов – Гидрокортизона, Преднизолона.
- На данном этапе эффективны физиотерапевтические процедуры. Назначаются сеансы фонофореза с использованием стероидного гормона – Гидрокортизона.
Для симптоматического лечения при артритах назначаются хондропротекторы. Структум, Терафлекс, Артра препятствуют разрушению сустава, останавливают дегенеративные процессы в хрящевой ткани.
Для облегчения состояния глаз капают антигистаминные капли – Аллергодил, Лекролин, Алломид. Глазные средства снимают покраснения, устраняют зуд и слезотечение.

Профилактика
К мерам предупреждения болезни можно отнести:
- не вести развратную половую жизнь, соблюдать гигиену
- своевременное лечение заболеваний мочеполовой системы;
- обязательное лечение обоих партнеров при наличии венерического заболевания у одного из них.
На ранних этапах болезни Рейтера выздоровление наступает быстро без последствий для организма. Если подключаются аутоиммунные процессы, хроническое течение характеризуется присутствием остаточных явлений, которые находятся в стадии ремиссии.
При отсутствии лечения или неправильных действиях возможен летальный исход, поэтому обращение к специалисту необходимо при появлении первых симптомов со стороны любого органа.
Долгое время существовали две противоположные концепции на природу синдрома Рихтера. Согласно одной из них, синдром Рихтера представляет собой сочетание двух генетически не связанных друг с другом заболеваний. В клинической практике эта гипотеза находит прямое подтверждение только в тех исключительных случаях, когда течение В-клеточной лимфатической опухоли осложняется присоединением крупноклеточной лимфомы с иным (Т-клеточным) иммунологическим фенотипом.
Сложнее трактовка наблюдений, когда обе болезни имеют одинаковую, реже Т-, а чаще В-линейную, принадлежность.
В тех случаях, когда на мембране клеток лимфоцитарной и крупноклеточной опухолей обнаруживают иммуноглобулины, идентичные по типу Н- и L-цепей, кажется очевидным, что обе болезни развиваются из одного исходного клона. Иногда экспрессия идентичных по типу L-цепей может сочетаться с разными изотипами (классами) иммуноглобулинов. Такие находки тоже скорее подтверждают, чем опровергают, идею о клональной прогрессии при синдроме Рихтера, поскольку в отличие от лимфоцитарной лимфомы/хронического лимфолейкоза на опухолевых элементах крупноклеточной лимфомы обнаруживались изотипы, свойственные более поздним этапам иммунного ответа —(М + D) => М => G => А (феномен переключения изотипов Н-цепей).
Если на мембране лимфоцитов и крупных опухолевых элементов определяются иммуноглобулины одного класса, но с разным типом L-цепей, например Igrvk—IgMA или IgMh—IgMk, предполагают, что заболевания происходят из разных клонов опухолевых клеток.
Установлено, что каждая полипептидная цепь иммуноглобулина кодируется несколькими генетическим элементами, которые в зародышевой конфигурации пространственно разобщены. В предшественниках В-лимфоцитов эти элементы в результате рекомбинации ДНК должны расположиться рядом, чтобы образовался единый активный генный комплекс, способный кодировать синтез тяжелых (VHD-JH + СH) или легких (VLJL + CL) полипептидных цепей.
Иными словами, в результате соматической перестройки молекулы ДНК происходит объединение рассредоточенных (V1=>n — variable, D1-5 — diversity, J1-4 — joining, Cu,q,y,a,e — constat) генных сегментов, и из множества вариантов производится сборка одного, уникального для данной популяции клеток кло-нального маркера. Этот процесс, обозначаемый как реаранжировка генов иммуноглобулинов, приводит к образованию фрагмента ДНК, отличающегося от зародышевой конфигурации.
Образец перестройки (реаранжировки) генов тяжелых и к или X легких цепей является уникальным в каждой конкретной В-клеточной опухоли. В опухоли при этом имеются множественные копии идентичных V(D).1-объединений, отражающих принадлежность клеток к одному клону. При синдроме Рихтера анализ реаранжировки генов иммуноглобулинов методом блоттинга по Саузерну используется как важный дополнительный молекулярно-биологический способ изучения клональной связи между двумя различными по своим морфологическим характеристикам популяциями опухолевых клеток.
Метод блот-гибридизации позволяет не только выявить рестрикционные фрагменты ДНК, отражающие генные объединения V(D)J в лимфоцитах крови/костного мозга и клетках экстрамедуллярной опухолевой ткани, но и сравнивать их. Обнаружение одинаковых клональных полос реаранжировки генов Н- и/или L-цепей дает основание полагать, что синдром Рихтера чаще всего представляет собой моноклоновый злокачественный процесс. Обычно в таких случаях обе популяции опухолевых клеток экспрессируют на своей поверхности только один тип L-цепей — либо к, либо X. В тех случаях, когда в лимфоцитах и крупных опухолевых элементах гены иммуноглобулинов перестроены по-разному, утверждается, что заболевания при синдроме Рихтера не имеют клональной связи друг с другом. В этих случаях опухоли, как правило, отличаются по типу синтезируемых L-цепей.
Затруднительными для интерпретации представляются ситуации, при которых из двух типов перестройки генов иммуноглобулинов, присутствующих в опухоли, только один совпадает с образцом реаранжировки в В-лимфоцитах крови/костного мозга. Серьезные затруднения в трактовке могут возникать, если результаты генных исследований вступают в противоречие с данными, полученными при иммунологическом фенотипировании опухолевых клеток. Подобная ситуация описана К. Miyamura и соавт..
В публикации речь идет о пациенте 71 года с лимфоцитозом крови/костного мозга, шейной лимфаденопатией, гепато- и спленомегалией и симптомами общей интоксикации. Морфологически в препаратах лимфатического узла обнаружена картина диффузной крупноклеточной лимфомы.
В представленном наблюдении клетки лимфоцитарной и крупноклеточной опухолей, с одной стороны, имели идентичный иммунофенотип (CD5+CD19+CD20+HLA-DR+) и одинаковые реаранжировки генов IgH, с другой — они различались по типу экспрессируемых L-цепей — X и к соответственно. Последнее обстоятельство не помешало авторам сделать вывод о том, что обе болезни, вероятнее всего, имели общее клональное происхождение, по крайней мере на начальных этапах малигнизации.
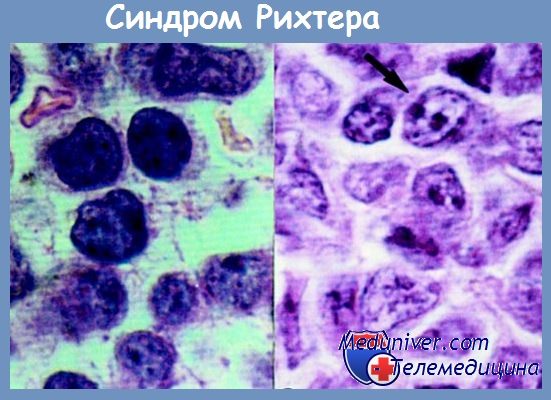
Для теоретического обоснования такого подхода предложены две гипотетические точки зрения. Согласно одной из них, онкогенное событие у данного больного могло произойти очень рано в ряду В-клеточной дифференцировки: после перестройки генов Н-цепей, но до реаранжировки генов L-цепей иммуноглобулинов. Другое объяснение единого происхождения двух заболеваний заключается в том, что исходный (лимфоцитарный) опухолевый клон имел мембранный IgMk. После клональной эволюции в крупноклеточную лимфому делеция к-генов и реаранжировка h-генов произошли только в популяции опухолевых лимфоцитов. Нам представляется вероятным и другое объяснение данного наблюдения: возможно, родоначальные клетки лимфоцитарной опухоли не делетировали к-гены и персистировали на протяжении болезни в «минорных», не улавливаемых методом Саузерна количествах.
В дальнейшем, вероятно, именно этот клон наиболее эффективно трансформировался в крупноклеточную лимфому.
Длительное время было неясно, насколько лимфоциты лимфоцитарной лимфомы/хронического лимфолейкоза являются функционально активными и способными к дальнейшей дифференцировке. Решению этой проблемы в определенной степени способствовала работа L. F. Bertoli и соавт., в которой, кроме того, продемонстрирован иной подход к изучению клональных взаимоотношений при синдроме Рихтера. Авторы получили моноклональные антитела к идиотипической (антигенной) детерминанте молекул Ig(M+D) Х-типа, экспрессированных на лимфоцитах больной хроническим лимфолейкозом.
В двухцветной иммунофлюоресценции В-клетки с мембранным Ig(M+D)A. идентифицировались с помощью антиидиотипических антител как преобладающая в лейкемическом клоне клеточная популяция. Вместе с тем было показано, что в определенной части IgG и IgA В-клеток, а также в большинстве IgG плазмоцитов костного мозга и крови обнаруживалась идиотипическая детерминанта, аналогичная таковой в лейкемических В-лимфоцитах. Следовательно, лейкозные лимфоциты или по крайней мере их некоторая часть способны к восприятию антигенных стимулов, изотипическому переключению и дифференцировке в плазматические клетки.
Через 6 лет после установления диагноза хронического лимфолейкоза больная умерла при явлениях дыхательной недостаточности, обусловленной опухолевой инфильтрацией легочной ткани. На вскрытии выявлена диффузная крупноклеточная лимфома, клетки которой экспрессировали на своей поверхности IgMh с таким же идиотипом, как и лимфоциты исходного клона. Кроме того, при блоттинге по Саузерну в лейкемических клетках крови и ткани лимфомы были получены идентичные клональные полосы перестройки генов Н-цепей иммуноглобулинов.
Таким образом, в данном наблюдении общее клональное происхождение хронического лимфолейкоза и диффузной крупноклеточной лимфомы было подтверждено не только при изучении иммунофенотипа, генных реаранжировок, но и с помощью антиидиотипических антител к молекулам иммуноглобулинов, экспрессированным на опухолевых клетках. В результате этих исследований одновременно получены косвенные доказательства способности В-лимфоцитов хронического лимфолейкоза к дифференцировке вплоть до плазмоцитов на протяжении самой болезни. При блоке развития лейкемических клеток на более позднем этапе дифференцировки, по-видимому, возможна трансформация в крупноклеточную лимфому.
Подобная возможность подтверждается наблюдением Е. Cofrancesco и соавт.. У больной с 6-летним анамнезом хронического лимфолейкоза развилась крупноклеточная лимфома с генерализованным поражением лимфатических узлов, печени, селезенки, кишечника, надпочечников, почек, костей. Обе популяции лимфоидных клеток (мелкие и крупные) имели идентичные реаранжировки генов иммуноглобулинов и сходные иммунофенотипические характеристики (CD5+CD19+CD20+HLA-DR+CD10). Однако крупные клетки лимфомы отличались от лимфоцитов (клеток лимфолейкоза) большей степенью иммунологической дифференцировки в направлении плазмоцитов. Это подтверждалось появлением цитоплазматических IgM, экспрессией CD38, утратой мембранных IgD и снижением розеткоообразования с мышиными эритроцитами.
При комбинированных В-клеточных опухолях клональная связь между злокачественными процессами изучали также посредством структурного анализа генов иммуноглобулинов. Работа V. Cherepa-khin и соавт. была первой, в которой по последовательности нуклеотидов ДНК показано, что клетки крупноклеточной лимфомы и хронического лимфолейкоза при синдроме Рихтера могут происходить из одного клона, несмотря на разный иммунофенотип: клетки крупноклеточной опухоли у больного были CD5-негативными.
J. Seymour и J. Campbell проанализировали известные исследованные современными методами случаи синдрома Рихтера. Они установили, что примерно в 2/3 случаев хронический лимфолейкоз и развившаяся крупноклеточная лимфома происходят из одного клона, в 1/3 — из разных.
Гематологам известно, что далеко не всегда при наличии зрелоклеточной лимфатической опухоли развивается крупноклеточная лимфома. Более того, синдром Рихтера — это редкий клинико-морфологический феномен. По данным различных авторов, он встречается только у 3—10 % больных лимфоцитарной лимфомой/хроническим лимфолейкозом. В такой ситуации понятны попытки выяснить, чем основная масса лимфоцитарных опухолей отличается от той небольшой части, которая осложняется развитием крупноклеточной лимфомы. Выдвигается и обосновывается довольно логичное предположение о возможном существовании подгруппы В-клеточных лимфатических опухолей, более подверженных внешним воздействиям и/или неконтролируемой бласттрансформации.
Подтверждением этому может служить обнаружение в лимфоцитах при ХЛЛ, осложненном развитием крупноклеточной лимфомы, замещающей мутации в D- и/или Jн-сегментах генов иммуноглобулинов. Большинство же случаев лимфоцитарной лимфомы/ хронического лимфолейкоза обычно представлено популяцией В-лимфоцитов, не имеющих таких мутаций. Результаты изучения реаранжировки протоонкогенов BCL-1, BCL-2, c-MYC, некоторых супрессорных генов, а также ТР53 иногда представляют большую сложность для интерпретации. Например, при различной реаранжировке генов иммуноглобулинов в опухолевых клетках при лимфолейкозе и крупноклеточной лимфоме могут обнаруживаться идентичные перестройки протоонкогена BCL-2.
Пока до конца неясны механизмы развития крупноклеточной опухоли в тех случаях, когда она происходит из того же клеточного клона, что и хронический лимфолейкоз. Изучение влияния ростовых факторов (TGF-p, G-CSF), цитогенетических изменений, мутаций генов ТР53 и р16, чаще наблюдающихся при синдроме Рихтера, чем при ХЛЛ, c-MYC, реаранжировка которого обнаружена в некоторых случаях синдрома Рихтера, не внесло определенности.
Много работ посвящено изучению роли вируса Эпштейна — Барр в развитии синдрома Рихтера. Показано, что ДНК вируса обнаружена только при развитии крупноклеточной лимфомы с клетками, напоминающими клетки Рид — Штернберга, и крупноклеточной опухоли с Т-клеточным иммунофенотипом. Во всех этих случаях доказано, что лимфоциты ХЛЛ и клетки лимфомы происходят из разных клонов.
Неясной остается роль иммуносупрессии, в частности, вызываемой лечением флударабином. В некоторых сообщениях, основанных на небольшом количестве наблюдений, показано определенное увеличение частоты синдрома Рихтера у больных ХЛЛ, получавших флударабин. В больших сериях, основанных на наблюдениях нескольких сотен длительно прослеженных больных, увеличения синдрома Рихтера у получавших пуриновые аналоги не отмечено. Возможно, указанное некоторыми авторами увеличение частоты крупноклеточной лимфомы при лечении флударабином объясняется более агрессивным течением ХЛЛ у этих больных, что и потребовало применения флударабина.
Развитие крупноклеточной лимфомы у больных со зрелоклеточными лимфопролиферативными заболеваниями является плохим в прогностическом отношении признаком и, как правило, сопровождается появлением ряда новых клинических симптомов. Чаще всего наблюдаются следующие:
1) резкое увеличение лимфатических узлов, особенно брюшной полости, значительное, иногда изолированное поражение селезенки и/или появление изолированных экстранодальных опухолевых образований;
2) появление общих симптомов: повышение температуры тела до 38 С и более без видимой инфекционной причины, снижение массы тела, выраженная потливость;
3) повышение уровня ЛДГ;
4) гиперкальциемия;
5) регрессия лимфоцитоза крови и/или костного мозга, совпадающая, а иногда предшествующая по времени появлению общих симптомов и генерализации экстрамедуллярного опухолевого процесса;
6) резкое ухудшение состояния больного.
Продолжительность жизни после обнаружения крупноклеточной опухоли обычно колеблется от 6 до 12 мес, несмотря на применение адекватных при лимфомах высокой степени злокачественности методов комбинированной химиотерапии.
При синдроме Рихтера иногда наблюдается изолированная экстранодальная локализация очагов крупноклеточной лимфомы. Так, описано поражение кожи, мягких тканей с прорастанием в позвонок и его деструкцией, вещества мозга, яичек, желудка и/или кишечника, бронхиального дерева с эндобронхиальным ростом опухоли. Появление перечисленных признаков у больных с лимфоцитарной опухолью должно служить основанием для проведения диагностической биопсии.
В последующем необходимо иммуноморфологическое исследование опухолевой ткани с обязательным цитологическим изучением отпечатков для исключения синдрома Рихтера.
Мы наблюдали 13 больных со злокачественными лимфопролиферативными заболеваниями, протекавшими с лимфоцитозом крови и костного мозга, у которых развилась крупноклеточная лимфома. Эти больные составили 3,2 % от общего числа больных с периферическими мелкоклеточными В-лимфомами. Среди больных с синдромом Рихтера было 6 женщин и 7 мужчин в возрасте от 40 до 77 лет.
Основным клиническим проявлением злокачественного процесса у 12 больных было увеличение лимфатических узлов разных групп с лимфоцитозом крови и костного мозга. У одного пациента экстрамедуллярный компонент лимфоцитарной опухоли характеризовался изолированным поражением селезенки.
В морфологическом субстрате у всех больных преобладали малые лимфоциты. Субстрат лимфоцитарной опухоли характеризовался типичной коэкспрессией зрелыми В-лимфоцитами маркеров CD5 и CD23. Опухолевые элементы крупноклеточной В-лимфомы отличались иммунофенотипически от субстрата лимфоцитарной лимфомы отсутствием экспрессии CD5 во всех, a CD23 — в половине исследованных случаев.
Через 8—180 мес (медиана 65 мес) течение зрелоклеточного лимфопролиферативного процесса осложнилось развитием крупноклеточной лимфомы с поражением лимфатических узлов и/или экстранодальной локализацией очагов опухолевого роста. У разных больных наблюдалось поражение различных органов и тканей: кожи, мягких тканей, костей, молочной железы, сальника, плевры с развитием плеврита, носоглотки. Подобная «трансформация» у 8 больных сопровождалась ухудшением состояния, причем у четырех отмечались общие симптомы. У остальных пациентов самочувствие оставалось без изменений. В период развития крупноклеточной (иммунобластной) лимфомы у 5 из 13 больных наблюдалась самопроизвольная регрессия лимфоцито-за крови и костного мозга, т. е. исчезновение основного признака лимфоцитарной опухоли. У 2 больных, напротив, генерализация иммунобластной лим-фомы сопровождалась ростом лимфоцитоза крови и костного мозга до самых высоких значений за весь период наблюдения.
Продолжительность жизни после установления диагноза крупноклеточной лимфомы широко варьировала — от 1 до 106 мес (медиана 8 мес).