
Анализ на адреногенитальный синдром
Содержание:
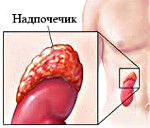
Для данной патологии характерны отклонения в строении и функционировании половых органов. Происхождение заболевания пока не установлено, однако медики считают, что синдром развивается вследствие чрезмерной выработки корой надпочечников андрогенов. Болезнь может быть вызвана различными опухолями или врожденной гиперплазией желез.
Что такое адреногенитальный синдром
Врождённая гиперплазия надпочечников является самым распространенным типом верулизующих патологий. Адреногенитальный синдром — это заболевание, которое известно мировой медицине, как синдром Апера-Гаме. Его развитие связано с повышенной выработкой андрогенов и выраженным снижением уровня кортизола и альдостерона, причиной чему служит врожденная дисфункция коры надпочечников.
Последствия отклонения могут быть серьезными для новорожденного, поскольку кора надпочечников отвечает за производство огромного количества гормонов, которые регулируют работу большинства систем организма. В результате патологии в теле ребенка (это может наблюдаться как у мальчиков, так и у девочек) становится слишком много мужских гормонов и очень мало женских.
Тип наследования
Каждая форма заболевания связана с генетическими нарушениями: как правило аномалии имеют наследственную природу и переходят от обоих родителей к ребенку. Более редки случаи, когда тип наследования адреногенитального синдрома является спорадическим – возникает внезапно в период формирования яйцеклетки или сперматозоида. Наследование адреногенитального синдрома происходит аутосомно-рецессивным путем (когда оба родителя являются носителями патологического гена). Иногда болезнь поражает детей в здоровых семьях.
Адреногенитальный синдром (АГС) характеризуется следующими закономерностями, влияющими на вероятность поражения им ребенка:
- если родители здоровы, но оба являются носителями гена StAR дефицита, есть риск, что новорожденный будет болеть врожденной гиперплазией надпочечников;
- если у женщины или мужчины диагностировали синдром, а второй партнер имеет нормальную генетику, то все дети в их семье будут здоровыми, однако станут носителями заболевания;
- если один из родителей болен, а второй является носителем адреногенетической патологии, то половина детей в данной семье будут болеть, а другая половина – носить мутацию в организме;
- при наличии болезни у обоих родителей, все их дети будут иметь аналогичные отклонения.

Формы
Андрогенетическое заболевание условно подразделяется на три типа – вирильный простой, сольтеряющий и постпубертатный (неклассический). Разновидности имеют серьезные отличия, поэтому каждый пациент требует детальной диагностики. Как проявляются формы адреногенитального синдрома:
- Вирильная форма. Для нее характерно отсутствие признаков наличия надпочечниковой недостаточности. Остальные симптомы АГС присутствуют в полном объеме. Данный тип патологии крайне редко диагностируют у новорожденных, чаще – у подростков (юношей и девушек).
- Сольтеряющий тип. Диагностируется исключительно у младенцев в течение первых недель/месяцев жизни. У девочек наблюдается псевдогермафродитизм (наружные половы органы схожи с мужскими, а внутренние – женские). У мальчиков сольтеряющий синдром выражается так: половой член имеет непропорционально большой размер относительно тельца, а кожный покров мошонки имеет специфическую пигментацию.
- Неклассический вид. Для патологии характерно наличие неясной симптоматики и отсутствие выраженной дисфункции надпочечников, что сильно усложняет диагностику АГС.
Адреногенитальный синдром — причины
Врожденная дисфункция надпочечников объясняется лишь проявлением наследственной болезни, поэтому приобрести в течение жизни или заразиться такой патологией невозможно. Как правило, проявляется синдром у новорожденных малышей, но редко АГС диагностируют у молодых людей возрастом до 35 лет. При этом активизировать механизм патологии могут такие факторы, как прием сильнодействующих препаратов, повышенный радиационный фон, побочное действие от гормональных контрацептивов.
Каким бы ни был стимул для развития болезни, причины адреногенитального синдрома носят наследственный характер. Прогноз выглядит примерно так:
- если в семье хотя бы 1 родитель здоров, ребенок вероятно родится без патологии;
- у пары, где один носитель, а другой болен АГС в 75% случаев родится больной ребенок;
- у носителей гена риск иметь больного ребенка равен 25%.

Симптомы
АГС не относится к числу смертельно опасных болезней, однако некоторые его симптомы доставляют человеку серьезные психологические неудобства и нередко приводят к нервному срыву. При диагностике патологии у новорожденного родители имеют время и возможность помочь ребенку с социальной адаптацией, а если заболевание обнаруживается в школьном возрасте или позже, ситуация может выйти из-под контроля.
Установить наличие АГС можно исключительно после проведения молекулярно-генетического анализа. Симптомы адреногенитального синдрома, которые указывают на необходимость диагностики — это:
- нестандартная пигментация кожных покровов ребенка;
- устойчивый рост артериального давления;
- несоответствующий возрасту ребенка низкий рост (из-за быстрого окончания выработки соответствующего гормона рано происходит остановка роста);
- периодические судороги;
- проблемы с пищеварением: рвота, поносы, сильное газообразование;
- у девочек половые губы, клитор недоразвиты или же, наоборот, имеют увеличенные размеры;
- у мальчиков внешние половые органы имеют непропорционально большие размеры;
- у девушек с АГС наблюдаются проблемы с менструациями, зачатием ребенка (часто сопутствует болезни бесплодие), вынашиванием плода;
- у пациенток женского пола часто происходит оволосение половых органов по мужскому типу, кроме того, наблюдается рост усов, бороды.
Адреногенитальный синдром у новорожденных
Заболевание может быть обнаружено на ранней стадии у новорожденных детей, что связано с проведением неонатального скрининга на 4 сутки после рождения ребенка. В ходе теста каплю крови из пятки малыша наносят на тест-полоску: если реакция положительная, ребенка переводят в эндокринологический диспансер и проводят повторную диагностику. После подтверждения диагноза начинается лечение АГС. Если адреногенитальный синдром у новорожденных обнаружен рано, то терапия проходит легко, в случаях позднего обнаружения адреногенетической патологии сложность лечения растет.

У мальчиков
Болезнь у детей мужского пола развивается, как правило, с двухлетнего или трехлетнего возраста. Происходит усиленное физическое развитие: увеличиваются гениталии, осуществляется активное оволосение, начинают появляться эрекции. При этом яички отстают в росте, а в дальнейшем вовсе прекращают развиваться. Как и у девочек, адреногенитальный синдром у мальчиков характеризуется активным ростом, однако он длится недолго и в итоге человек все равно остается низким, коренастым.
У девочек
Патология у девочек зачастую выражается сразу при рождении в вирильной форме. Ложный женский гермафродитизм, характерный для АГС, характеризуется увеличенным размером клитора, при этом отверстие мочеиспускательного канала находится прямо под его основанием. Половые губы в данном случае напоминают по форме расщепленную мужскую мошонку (урогенитальный синус не делится на влагалище и уретру, а останавливается в развитии и открывается под пенисообразным клитором).
Не редко адреногенитальный синдром у девочек так ярко выражен, что при рождении младенца трудно сразу установить его пол. В период 3-6 лет у ребенка активно растут волосы на ногах, лобке, спине и девочка внешне становится очень похожа на мальчика. Больные АГС дети растут гораздо быстрее своих здоровых сверстников, однако их половое развитие вскоре полностью прекращается. При этом молочные железы остаются маленькими, а менструации либо полностью отсутствуют, или же появляются нерегулярно из-за того, что недоразвитые яичники не могут в полном объеме выполнять свои функции.
Диагностика адреногенитального синдрома
Выявить заболевание можно при помощи современных исследований гормонального фона и при визуальном осмотре. Врач при этом учитывает анамнестические и фенотипические данные, к примеру, оволосение в нетипичных для женщины местах, развитость молочных желез, мужской тип телосложения, общий вид/здоровье кожи, пр. АГС развивается вследствие недостаточности 17-альфа-гидроксилазы, поэтому в крови пациента можно отследить уровень гормонов ДЭА-С и ДЭА, являющихся предшественниками тестостерона.
Диагностика также включает анализ мочи на определение показателя 17-КС. Биохимический анализ крови позволяет установить уровень гормонов 17-ОНП и ДЭА-С в теле пациента. Комплексная диагностика, кроме того, подразумевает изучение симптоматики гиперандрогении и прочие нарушения работы эндокринной системы. При этом показатели проверяют дважды – до пробы с глюкокортикостероидами и после нее. Если во время анализа уровень гормонов сокращается до 75% или больше процентов – это говорит о выработке андрогенов исключительно корой надпочечников.
Кроме анализов на гормоны, диагностика адреногенитального синдрома включает УЗИ яичников, при котором врач определяет ановуляцию (ее можно выявить, если наблюдаются фолликулы разного уровня зрелости, не превышающие преовуляторных объемов). В таких случаях яичники имеют увеличенные размеры, но объем стромы находится в норме и отсутствуют фолликулы под капсулой органов. Только после проведения развернутого обследования и подтверждения диагноза начинается лечение адреногенитального синдрома.

Адреногенитальный синдром – лечение
АВС – это не фатальная патология с летальным исходом, поэтому вероятность развития необратимых изменений в организме больного крайне мала. Тем не менее современное лечение адреногенитального синдрома не может похвастаться своей эффективностью и действенностью. Пациенты с таким диагнозом вынуждены пожизненно принимать гормональные препараты, чтобы восполнять дефицит гормонов группы глюкокортикостероидов, и бороться с чувством неполноценности.
Пока остаются неизученными перспективы подобной терапии, но есть данные, которые свидетельствуют о высокой вероятности развития сопутствующих АГС патологий сердца, костей, сосудов, органов ЖКТ, онкологических болезней. Этим объясняется необходимость проводить людям с дисфункцией коры надпочечников регулярные обследования – делать рентген костей, электрокардиограмму, УЗИ брюшины, пр.
OMIM 201910
Наша команда профессионалов ответит на ваши вопросы
Адреногенитальный синдром (АГС) – врожденное заболевание, характеризующееся прогрессирующей вирилизацией, ускоренным соматическим развитием, повышенной экскрецией гормонов коры надпочечников. Наиболее часто встречается тип гиперплазии надпочечников, вызванный дефицитом 21-гидроксилазы. Дефицит 21-гидроксилазы приводит к нарушению образования дезоксикортикостерона и 11-дезоксикортизола. Биосинтез андрогенов не нарушен, что приводит к их избыточной продукции еще при внутриутробном развитии. У новорожденных девочек отмечается разная степень маскулинизации от умеренной гипертрофии клитора до полного срастания губно-мошоночных складок с формированием предстательной железы, мошонки, полового члена с отверстием мочеиспускательного канала. Внутренние половые органы сформированы правильно, кариотип 46,ХХ. Отмечается гиперпигментация околососковой и генитальной областей. У мальчиков основными клиническими симптомами являются раннее половое развитие и низкий рост, связанный с преждевременным закрытием зон роста эпифизов.
Для вирильной формы адреногенитального синдрома, составляющей 1/3 случаев, характерны данные признаки, электролитный баланс, уровень кортизола и альдостерона в норме.
Сольтеряющая форма составляет 2/3 классических случаев адреногенитального синдрома. Кроме вирилизующего действия избытка андрогенов наблюдается нарушение солевого обмена в результате дефицита минералокортикоидов и вовлечения в патологический процесс ренин-альдостероновой системы. Заболевание проявляется в первые дни жизни срыгиванием, рвотой, потерей веса, сонливостью, признаками дегидротации, гипонатриемией, гиперкалиемией, метаболическим ацидозом.
При поздней (неклассической) форме адреногенитального синдрома отсутствуют симптомы вирилизации у девочек при рождении. Первые признаки появляются в подростковом возрасте. Для девочек характерно умеренное увеличение клитора, ускорение костного возраста, нарушение менструальной функции, маскулинное телосложение. У мальчиков признаки менее заметны, единственными симптомами могут быть ускорение костного возраста и преждевременное половое оволосение. Рост взрослых ниже среднего, фертильность снижена.
Во всех случаях заболевания отмечается повышение в моче уровня 17-кетостероидов и прегнантриола.
Тип наследования – аутосомно-рецессивный.
Популяционная частота – 1:1 000 000 неклассическая форма, 1:18 000 – классическая форма.
Ген, мутации в котором приводят к развитию АГС, картирован на хромосоме 6р21.3. Ген 21-гидроксилазы (CYP21OHB, CYP21А2) и псевдоген 21-гидроксилазы (CYP21OHA) организованы в тандем повторов с генами С4В и С4А комплемента между генами HLA-B и HLA-DR. Из-за высокой степени гомологии между повторами неравные рекомбинации и генная конверсия ответственны за большинство мутаций в этом локусе. Ген CYP21A2 состоит из 10 экзонов, имеет длину 6.3 т.п.н. Ген и псевдоген гомологичны на 98%. Гипотетический продукт псевдогена полностью не функционален. 30% обнаруженных мутаций составляет делеция гена CYP21A2. Эта мутация ответственна за развитие сольтеряющей формы заболевания. Другой мутацией, ответственной за развитие сольтеряющей формы, является мутация сайта сплайсинга во втором интроне (35%). Большинство из других мутаций в норме присутствует в псевдогене CYP21OHA и, вероятно, представляют собой результат генной конверсии. К сольтеряющей форме заболевания приводят мутации Q318X, кластер миссенс-мутаций в экзоне 6 (I235N/V236E/ M238K). Для вирильной формы характерна мутация I172N. За развитие неклассической формы заболевания с поздним началом ответственны, в большинстве случаев, мутации P30L, V281L, P453S. В Западной Европе 60% мутаций при неклассической форме АГС составляет мутация V281L.
В Центре молекулярной генетики проводится поиск 9 частых мутаций: делеции гена CYP21А2, мутации сайта сплайсинга в интроне 2, мутаций P30L, I172N, I235N/V236E/M238K, V281L, Q318X, R356W, P453S. Данный анализ проводится для больных детей с обязательным предоставленим материала их родителей. материал родителей может понадобиться для исследования т.к. при конверсии больших участков псевдогена последовательность гена CYP21А2 может включать в себя несколько мутаций. Поэтому при проведении анализа необходимо определить, находятся ли мутации на одной хромосоме или лежат на разных хромосомах. Наличие материала родителей поможет в этих случаях определить расположение мутаций на хромосомах. Также данный вид поиска мутаций возможен для родителей при отсутствии материала от больного ребенка.
Для проведения поиска частых мутаций в гене CYP21А2 (CYP21OHB) необходимо предоставить свежую кровь, взятую в пробирку с консервантом ЭДТА.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий — около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Адреногенитальный синдром — наследственное заболевание надпочечников, при котором вследствие функциональной несостоятельности ферментов нарушается стероидогенез. Проявляется вирилизацией гениталий, маскулиноподобным телосложением, недоразвитием груди, гирсутизмом, акне, аменореей или олигоменореей, бесплодием. В ходе диагностики определяют уровни 17-гидроксипрогестерона, 17-кетостероидов, андростендиона, АКТГ, проводят УЗИ яичников. Пациенткам назначают заместительную гормонотерапию глюкокортикоидами и минералокортикоидами, эстрогены в комбинации с андрогенами или прогестинами нового поколения. При необходимости выполняют пластику половых органов.
МКБ-10
Общие сведения
Адреногенитальный синдром, или врожденная дисфункция (гиперплазия) коры надпочечников, — наиболее частое из наследуемых заболеваний. Распространенность патологии отличается у представителей разных национальностей. Классические варианты АГС у лиц европеоидной расы встречаются с частотой 1:14 000 младенцев, в то время как у эскимосов Аляски этот показатель составляет 1:282. Существенно выше заболеваемость у евреев. Так, неклассическую форму адреногенитального расстройства выявляют у 19% лиц еврейской национальности группы ашкенази. Патология передается по аутосомно-рецессивному типу. Вероятность рождения ребенка с таким синдромом при носительстве патологического гена у обоих родителей достигает 25%, в браке носителя и больного — 75%. Если один из родителей имеет полноценные ДНК, клинические проявления синдрома у детей не развиваются. При наличии АДС у отца и матери ребенок также будет болен.
Причины адреногенитального синдрома
У больных с наследуемой гиперплазией надпочечников генетический дефект проявляется несостоятельностью ферментных систем, участвующих в секреции стероидных гормонов. В 90-95% случаев патология возникает при повреждении гена, который отвечает за синтез 21-гидроксилазы — фермента, влияющего на образование кортизола. В остальных клинических случаях вследствие дефекта ДНК нарушается производство других ферментов, обеспечивающих стероидогенез, — StAR/20,22-десмолазы, 3-β-гидрокси-стероиддегидрогеназы, 17-α-гидроксилазы/17,20-лиазы, 11-β-гидроксилазы, P450-оксидоредуктазы и синтетазы альдостерона.
У пациентов с признаками вирилизирующего синдрома вместо активного гена CYP21-B в коротком плече 6-й аутосомы расположен функционально несостоятельный псевдоген CYP21-A. Структура этих участков ДНК-цепи во многом гомологична, что повышает вероятность конверсии генов в мейозе с перемещением участка нормального гена на псевдоген или делецию CYP21-B. По-видимому, именно этими механизмами объясняется существование скрытых форм болезни, дебютирующих в пубертате или постпубертатном периоде. В таких случаях клинические признаки патологии становятся заметными после нагрузок, истощающих кору надпочечников: тяжелых болезней, травм, отравлений, радиационных воздействий, длительного периода интенсивной работы, психологически напряженных ситуаций и т. д.
Патогенез ВДКН
В основе механизма развития наиболее распространенного варианта адреногенитального синдрома с дефектом CYP21-B-гена лежит принцип обратной связи. Ее начальным звеном становится дефицит стероидов — кортизола и альдостерона. Несостоятельность процессов гидроксилирования сопровождается неполным переходом 17-гидроксипрогестерона и прогестерона в 11-дезоксикортизол и дезоксикортикостерон. В результате снижается секреция кортизола, а для компенсации этого процесса в гипофизе усиливается синтез АКТГ — гормона, вызывающего компенсаторную гиперплазию коры надпочечников для стимуляции выработки кортикостероидов.
Параллельно возрастает синтез андрогенов и появляются видимые признаки их влияния на чувствительные ткани и органы. При умеренном снижении активности фермента минералокортикоидная недостаточность не развивается, поскольку потребность организма в альдостероне почти в 200 раз ниже по сравнению с кортизолом. Только глубокий дефект гена вызывает тяжелую клиническую симптоматику, которая проявляется с раннего возраста. Патогенез развития заболевания при нарушении структуры других участков ДНК аналогичен, однако пусковым моментом являются нарушения в других звеньях стероидогенеза.
Классификация
Систематизация различных форм вирилизирующей гиперплазии надпочечников основана на особенностях клинической картины заболевания, выраженности генетического дефекта и времени проявления первых патологических признаков. Тяжесть расстройства напрямую связана со степенью повреждения ДНК. Специалисты в сфере эндокринологии различают следующие виды адреногенитального синдрома:
- Сольтеряющий. Самый тяжелый вариант патологии, проявляющийся в первый год жизни ребенка грубыми нарушениями строения наружных половых органов у девочек и их увеличением у мальчиков. Активность 21-гидроксилазы составляет не более 1%. Значительное нарушение стероидогенеза приводит к выраженным соматическим нарушениям — рвоте, поносу, судорогам, чрезмерной пигментации кожи. Без лечения такие дети умирают в раннем возрасте.
- Простой вирильный. Течение заболевания менее тяжелое, чем при сольтеряющем варианте. Преобладают проявления неправильного развития гениталий у младенцев женского пола, увеличение их размеров у мальчиков. Признаки надпочечниковой недостаточности отсутствуют. Уровень активности 21-гидроксилазы снижен до 1-5%. С возрастом у пациентов нарастают признаки вирилизации вследствие стимулирующего действия андрогенов.
- Неклассический (постпубертатный). Наиболее благоприятная форма АГС, явные признаки которой возникают в период полового созревания и в репродуктивном возрасте. Наружные половые органы имеют нормальное строение, может быть увеличен клитор у женщин и половой член у мужчин. Функциональность 21-гидроксилазы снижена до 20-30%. Заболевание выявляется случайно при обследовании в связи с бесплодием или нарушениями менструальной функции.
Сольтеряющий и простой вирильный виды адреногенитальных расстройств относят к категории антенатальной патологии, формирующейся внутриутробно и проявляющейся с момента рождения. При дефекте строения других генов наблюдаются более редкие варианты заболевания: гипертензивные — классический (врожденный) и неклассический (поздний), гипертермический, липидный, с ведущими проявлениями гирсутизма.
Симптомы адреногенитального синдрома
При антенатальных формах заболевания (простой вирильной и сольтеряющей) основным клиническим симптомом является видимая вирилизация гениталий. У новорожденных девочек обнаруживаются признаки женского псевдогермафродитизма. Клитор большой по размерам или имеет пенисообразную форму, преддверие влагалища углублено, сформирован урогенитальный синус, большие и малые половые губы увеличены, промежность высокая. Внутренние половые органы развиты нормально. У младенцев-мальчиков увеличен половой член и гиперпигментирована мошонка. Кроме того, при сольтеряющем адреногенитальном расстройстве выражена симптоматика надпочечниковой недостаточности с тяжелыми, зачастую несовместимыми с жизнью соматическими нарушениями (понос, рвота, судороги, обезвоживание и др.), которые проявляются с 2-3-недельного возраста.
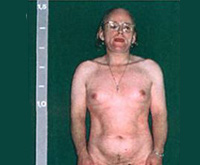
У девочек с простым вирильным АГС по мере взросления признаки вирилизации усиливаются, формируется диспластическое телосложение. Из-за ускорения процессов окостенения пациентки отличаются невысоким ростом, широкими плечами, узким тазом, короткими конечностями. Трубчатые кости массивные. Половое созревание начинается рано (до 7 лет) и протекает с развитием вторичных мужских половых признаков. Отмечается увеличение клитора, снижение тембра голоса, нарастание мышечной силы, формирование типичной для мужчин формы перстневидного хряща щитовидной железы. Грудь не растет, менархе отсутствует.
Менее специфичны клинические симптомы при неклассических формах вирилизирующего синдрома, возникшие в пубертате и после стрессовых нагрузок (выкидыша на ранних сроках беременности, медицинского аборта, операции и др.). Обычно пациентки вспоминают, что у них еще в младшем школьном возрасте появилось небольшое оволосение в подмышечных впадинах и на лобке. В последующем развились признаки гирсутизма с ростом стержневых волос над верхней губой, по белой линии живота, в области грудины, в сосково-ареолярной зоне. Женщины с АГС предъявляют жалобы на стойкую угревую сыпь, пористость и повышенную жирность кожи.
Менархе наступает поздно — к 15-16 годам. Менструальный цикл неустойчив, интервалы между менструациями достигают 35-45 дней и более. Кровянистые выделения во время месячных скудные. Молочные железы небольшие. Клитор несколько увеличен. Такие девушки и женщины могут иметь высокий рост, узкий таз, широкие плечи. По наблюдениям специалистов в сфере акушерства и гинекологии, чем позже развиваются адреногенитальные расстройства, тем менее заметны внешние признаки, характерные для мужчин, и тем чаще ведущим симптомом становится нарушение месячного цикла. При более редких генетических дефектах пациентки могут жаловаться на повышение артериального давления или, наоборот, гипотонию с низкой работоспособностью и частыми головными болями, гиперпигментацию кожи с минимальными симптомами вирилизации.
Осложнения
Основным осложнением адреногенитального синдрома, по поводу которого пациентки обращаются к акушерам-гинекологам, является стойкое бесплодие. Чем раньше проявилось заболевание, тем меньше вероятность забеременеть. При значительной ферментной недостаточности и клинических проявлениях простого вирилизирующего синдрома беременность вообще не наступает. У забеременевших пациенток с пубертатными и постпубертатными формами заболевания возникают самопроизвольные выкидыши на раннем сроке. В родах возможна функциональная истмико-цервикальная недостаточность. Такие женщины более склонны к возникновению психоэмоциональных расстройств — склонности к депрессии, суицидальному поведению, проявлениям агрессии.
Диагностика
Постановка диагноза при антенатальных типах АГС с характерными изменениями половых органов не представляет сложности и проводится сразу после родов. В сомнительных случаях применяют кариотипирование для подтверждения женского кариотипа (46ХХ). Большее значение диагностический поиск приобретает при позднем клиническом дебюте или скрытом течении с минимальными внешними проявлениями вирилизации. В подобных ситуациях для выявления адреногенитального синдрома используют следующие лабораторные и инструментальные методы:
- Уровень 17-ОН-прогестерона. Высокая концентрация 17-гидроксипрогестерона, который является предшественником кортизола — ключевой признак недостаточности 21-гидроксилазы. Его содержание увеличено в 3-9 раз (от 15 нмоль/л и выше).
- Стероидный профиль (17-КС). Повышение уровня 17-кетостероидов в моче у женщин в 6-8 раз свидетельствует о высоком содержании андрогенов, производимых корой надпочечников. При выполнении преднизолоновой пробы концентрация 17-КС уменьшается на 50-75%.
- Содержание андростендиона в сыворотке крови. Повышенные показатели этого высокоспецифичного метода лабораторной диагностики подтверждают усиленную секрецию предшественников мужских половых гормонов.
- Уровень АКТГ в крови. Для классических форм заболевания характерна компенсаторная гиперсекреция адренокортикотропного гормона передней долей гипофиза. Поэтому при синдроме вирилизирующей дисфункции показатель повышен.
- УЗИ яичников. В корковом веществе определяются фолликулы на разных стадиях созревания, не достигающие преовуляторных размеров. Яичники могут быть несколько увеличены, однако разрастания стромы не наблюдается.
- Измерение базальной температуры. Температурная кривая типична для ановуляторного цикла: первая фаза растянута, вторая укорочена, что обусловлено недостаточностью желтого тела, которое не образуется из-за отсутствия овуляции.
Для сольтеряющего варианта АГС также характерна повышенная концентрация ренина в плазме крови. Дифференциальная диагностика адреногенитальных расстройств, возникших в пубертатном и детородном возрасте, проводится с синдромом поликистозных яичников, овариальными андробластомами, андростеромами надпочечников, вирильным синдромом гипоталамического происхождения и конституциональным гирсутизмом. В сложных случаях к диагностике привлекают эндокринологов, урологов, врачей-генетиков.
Лечение адреногенитального синдрома
Основным способом коррекции вирильной дисфункции надпочечников является заместительная гормональная терапия, восполняющая дефицит глюкокортикоидов. Если у женщины со скрытым АГС нет репродуктивных планов, кожные проявления гиперандрогении незначительны и месячные ритмичны, гормоны не применяют. В остальных случаях выбор схемы лечения зависит от формы эндокринной патологии, ведущей симптоматики и степени ее выраженности. Зачастую назначение глюкокортикоидных препаратов дополняют другими медикаментозными и хирургическими методами, подобранными в соответствии с конкретной терапевтической целью:
- Лечение бесплодия. При наличии планов по деторождению женщина под контролем андрогенов крови принимает глюкокортикоиды до полного восстановления овуляторного месячного цикла и наступления беременности. В резистентных случаях дополнительно назначают стимуляторы овуляции. Во избежание выкидыша гормонотерапию продолжают до 13-й недели гестационного срока. В I триместре также рекомендованы эстрогены, во II-III — аналоги прогестерона, не обладающие андрогенным эффектом.
- Коррекция нерегулярных месячных и вирилизации. Если пациентка не планирует беременность, но жалуется на расстройство менструального цикла, гирсутизм, угри, предпочтительны средства с эстрогенным и антиандрогенным эффектом, оральные контрацептивы, содержащие гестагены последнего поколения. Терапевтический эффект достигается за 3-6 месяцев, однако по окончании лечения при отсутствии заместительной гормонотерапии признаки гиперандрогении восстанавливаются.
- Лечение врожденных форм АГС. Девочкам с признаками ложного гермафродитизма проводят адекватную гормонотерапию и выполняют хирургическую коррекцию формы половых органов — клитеротомию, интроитопластику (вскрытие урогенитального синуса). При сольтеряющих адреногенитальных расстройствах кроме глюкокортикоидов под контролем рениновой активности назначают минералокортикоиды с увеличением терапевтических доз при возникновении интеркуррентных заболеваний.
Определенные сложности в ведении пациентки возникают в тех случаях, когда заболевание не диагностировано в акушерском стационаре, и девочка с выраженной вирилизацией гениталий регистрируется и воспитывается как мальчик. При решении о восстановлении женской половой идентичности хирургическую пластику и гормонотерапию дополняют психотерапевтической поддержкой. Решение о сохранении гражданского мужского пола и удалении матки с придатками принимается в исключительных случаях по настоянию больных, однако такой подход считается ошибочным.
Прогноз и профилактика
Прогноз при своевременном обнаружении адреногенитального синдрома и адекватно подобранной терапии благоприятный. Даже у пациенток со значительной вирилизацией гениталий после пластической операции возможна нормальная половая жизнь и естественные роды. Заместительная гормонотерапия при любой форме АГС способствует быстрой феминизации — развитию грудных желез, появлению месячных, нормализации овариального цикла, восстановлению генеративной функции. Профилактика заболевания осуществляется на этапе планирования беременности.
Если в роду наблюдались случаи подобной патологии, показана консультация генетика. Проведение пробы с АКТГ обоим супругам позволяет диагностировать гетерозиготное носительство или скрытые формы адреногенитального расстройства. При беременности синдром может быть обнаружен по результатам генетического анализа клеток хорионических ворсин или содержимого околоплодных вод, полученных методом амниоцентеза. Неонатальный скрининг, проводимый на 5-е сутки после родов, направлен на выявление повышенной концентрации 17-гидропрогестерона для быстрого выбора терапевтической тактики.